
Brian J. Druker
Oregon Health & Science University

Nicholas B. Lydon
Novartis

Charles L. Sawyers
Memorial Sloan-Kettering Cancer Center
For the development of molecularly-targeted treatments for chronic myeloid leukemia, converting a fatal cancer into a manageable chronic condition.
The 2009 Lasker~DeBakey Clinical Medical Research Award honors three scientists who developed novel treatments for chronic myeloid leukemia (CML) that converted this fatal cancer into a manageable chronic condition. By targeting the molecular underpinnings of this disease, Brian J. Druker (Oregon Health & Science University), Nicholas B. Lydon (formerly, Novartis), and Charles L. Sawyers (Memorial Sloan-Kettering Cancer Center) broke new ground in cancer therapy and radically altered the prognosis of CML patients.
In the early phase of CML — known as the chronic stage — the body accumulates too many white blood cells, but these cells mature and function properly, and symptoms are not serious. Without treatment, the disease advances over a period of several years to a point of ‘blast crisis’, in which many immature blood and bone marrow cells accumulate — a condition that rapidly causes death. Few patients qualify for a bone marrow transplant to treat the disease, a risky prospect anyway, and, before the work of Druker, Lydon, and Sawyers, the rest were left with the drug of choice: interferon. This therapy prolongs survival by an average of only about two years and side effects are debilitating. Now, the five-year survival rate approaches 90 percent.
ABL to cause cancer
In 1960, Peter Nowell (Lasker Clinical Medical Research Award, 1998) and the late David Hungerford, working in Philadelphia, noticed an abnormally small chromosome in cells from patients with CML. The consistent presence of this so-called ‘Philadelphia chromosome’ prompted them to propose that the genetic lesion causes the disease. Thirteen years later, Janet Rowley (Lasker Clinical Medical Research Award, 1998), showed that the shortened chromosome — chromosome 22 — resulted from a swap between chromosomes 9 and 22.
Several world-class molecular biology groups then figured out that the Philadelphia chromosome generates an enzyme that promotes aberrant cell division. The chromosomal rearrangement places the tail of a gene called Abelson (ABL) from chromosome 9 onto the head of a gene called the breakpoint cluster region (BCR) from chromosome 22. The product of the resulting BCR-ABL fusion oncogene possesses ABL’s so-called tyrosine kinase activity — the ability to add phosphate chemical groups to the amino acid tyrosine — but fails to turn off appropriately. BCR-ABL’s continuous activity stimulates cell-growth pathways and transforms normal cells into ones that proliferate without restraint.
As these results emerged in the mid 1980s, Druker, who was training to be an oncologist at the Dana-Farber Cancer Institute, and Lydon, a biochemist at Ciba-Geigy (now Novartis) realized that blocking BCR-ABL might obliterate a CML cell’s ability to stir trouble. Until then, oncology drugs had relied on the fact that cancer cells reproduce faster than normal ones. Because chemotherapeutic agents targeted cell division in general, treatments killed not only cancer cells, but also healthy cells that must divide to perform their jobs. Perhaps by targeting BCR-ABL, a drug would strike only the misbehaving cells — the malignant ones — that depended on it to duplicate.
Although the idea held enormous appeal, many scientists thought it would not work. Hundreds of kinases operate in mammalian cells and any compound that quelled one would thwart them all, the skeptics argued, thus causing toxic side effects. Furthermore, although BCR-ABL by itself can cause CML, the diseased cells accumulate additional genetic flaws. Some of these anomalies might also spark cell division, in which case focusing on only the original one would not stifle disease.
By the early 1980s, the field of oncogenes had implicated unruly kinases in cancer, and Ciba-Geigy had begun to explore these proteins as potential drug targets. Lydon set up the company’s tyrosine kinase inhibitor program in 1986, under the direction of Alex Matter. Their team, which included cell biologist Elizabeth Buchdunger and chemist Jürg Zimmermann, was screening large chemical collections for compounds that hamper tyrosine kinases in test tubes and inside cells. They then chemically tweaked promising compounds, hoping to improve potency and selectivity. Along the way, Lydon began using a molecular tool that Druker made while studying kinases in the laboratory of Thomas Roberts (Dana-Farber Cancer Institute). This reagent, an antibody that detects the phosphotryosine product of the tyrosine kinase reaction, proved crucial to Lydon’s enzyme-activity measurements in cells.
In 1993, Druker set up his own laboratory with a single goal in mind: Find a company that had a BCR-ABL kinase inhibitor and develop it for clinical use in CML patients. He contacted Lydon, who sent several compounds for Druker to test. In 1996, Druker and Lydon reported that one of these substances, imatinib (now widely known as Gleevec), killed cultured cells that required BCR-ABL activity to survive, but did not affect a cell line that depended on a different tyrosine kinase, v-SRC. These same cell lines form tumors when injected into mice, and similarly, the compound quashed tumor formation of the BCR-ABL-producing cells, but not the v-SRC-producing cells. Patient samples provided especially exciting results. The compound did not harm normal cells, but it blocked multiplication of cells that carried BCR-ABL. Based on these studies, Gleevec emerged as the best compound to pursue.
Astonishing results
Ciba-Geigy and Druker assembled a team to design the clinical trials. A key member was Charles Sawyers, who was studying BCR-ABL at the University of California, Los Angeles. The study they conceived differed in several important ways from the norm. Crucially, rather than measuring only a clinical response — in this case, reducing white blood cell counts — they decided to track activity of BCR-ABL in remaining blood cells. Doing so would allow them to assess whether the drug was working as predicted — by blocking the enzyme’s activity. To perform the first clinical trials, they enlisted the collaboration of Moshe Talpaz, an oncologist at MD Anderson Cancer Center.
But before they could conduct the study, the enterprise hit some bumps. Ciba-Geigy merged with Sandoz to form Novartis, and Lydon left the company soon afterward. Toxicity concerns arose, although Druker thought they could easily be handled in the clinic. Passionate in his belief that Gleevec showed tremendous promise and eager to offer patients a potentially life-saving therapy, he lobbied Novartis to move the project forward.
Druker, Sawyers, and Talpaz finally got the go ahead to begin a clinical trial, which started in June 1998. The first study aimed primarily to assess Gleevec’s safety in chronic-phase CML patients who had not responded to interferon-based therapy or could not tolerate its side effects. Every month, a single patient at each institution began receiving the drug. As long as the patients experienced no severe side effects, a new set of three patients would receive the next higher dose the following month. At 300 milligrams, the patients’ white blood cell counts plunged over the first few weeks of therapy, returning to normal in 53 of the 54 patients treated with this or higher doses. Presumably, Gleevec was cutting off creation of new BCR-ABL-carrying white blood cells. Furthermore, the drug impeded activity of the enzyme in cells from patients — and in a third of the patients, the number of bone marrow cells with the Philadelphia chromosome plummeted after about six months of treatment. The patients experienced only mild side effects. These results were astonishing for a cancer drug. Typically, researchers hoped that about 10-20 percent of patients would respond in a clinical trial; in this study, 98 percent of the patients showed dramatic improvements.
Subsequent large-scale trials bore out the earlier results and, once the researchers knew the drug was working in chronic-phase patients, they offered it to those in blast crisis, the late stage of disease. At that point, Druker, Sawyers, and Talpaz saw something no oncologist had seen before. Patients on the edge of death were climbing out of bed and leaving the hospital within a week of their first Gleevec dose. In May of 2001, less than three years after the beginning of the first clinical study, the US Food and Drug Administration (FDA) approved the drug.
In the meantime, the researchers had organized an international study that compared interferon-based therapy with Gleevec in more than 1000 chronic-phase patients. After a year and a half, Gleevec was so outperforming interferon-based therapy that the researchers closed the trial and switched almost everyone to Gleevec. Five years after diagnosis, overall survival of patients treated with Gleevec was 89 percent, the researchers reported in 2006. The comparable statistic for interferon-treated patients (from other studies) was about 60 percent (see graph).
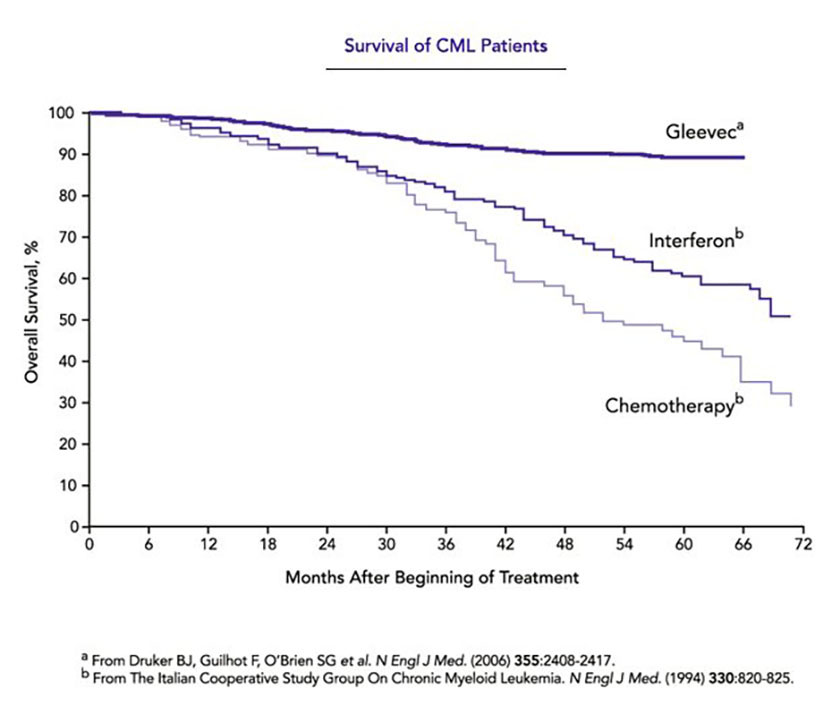
Survival of CML patients. Curves show overall survival for CML patients treated with Gleevec, interferon, or conventional chemotherapy. Conventional chemotherapy delivers only a minimal effect on survival compared with no treatment.
During the 2006 study, the scientists realized that Gleevec, as remarkable as it was, did not ‘cure’ patients in the strictest sense. Sensitive detection methods uncovered BCR-ABL-containing cells even when the usual laboratory tests did not reveal the Philadelphia chromosome, and individuals who discontinued Gleevec relapsed. Fortunately, because patients tolerate the drug well, they can take it long term.
Combating resistance
But another challenge was simmering. Some patients were developing resistance to the drug and Sawyers wanted to figure out why. Perhaps some substance in the bloodstream was soaking up the drug, or maybe CML cells no longer depended on BCR-ABL to duplicate, given the many other genetic perturbations in blast-stage cells. By assessing enzyme activity from patients over the course of their treatment, Sawyers discovered that a different scenario was at play. In patients who relapsed, Gleevec no longer dampened BCR-ABL activity: The enzyme itself had changed during therapy.
Initial analysis of the BCR-ABL gene revealed a sequence alteration that caused one amino acid to replace another at a particular spot in the protein. John Kuriyan (University of California, Berkeley) had recently deduced the structure of the BCR-ABL enzyme bound to Gleevec using X-ray crystallography, and the picture he produced explained what Sawyers observed. The amino acid change placed a large chemical group in the pocket where Gleevec normally grabs hold of the enzyme, which blocked the drug from binding.
Subsequent studies by Sawyers and others identified more than 50 genetic perturbations in BCR-ABL that confer resistance to Gleevec. Unlike the first mutation, they map mostly to locations other than points of contact between the drug and the enzyme. Many of these alterations likely prevent the enzyme from assuming its inactive form—the structure bound by Gleevec.
Sawyers proposed to find agents that block the resistant enzyme’s activity by fastening to BCR-ABL’s active form, yet barring it from performing its reaction. He pursued this tactic in record time with scientists at Bristol Myers Squibb to create a drug called Sprycel (desatinib) that binds BCR-ABL in its active as well as its inactive shape. Sprycel progressed rapidly through clinical trials and is FDA approved for patients with resistance to Gleevec. Scientists are exploring the possibility of treating people with combinations of Gleevec and second-generation medications such as Sprycel, with the hope of delaying or preventing the emergence of drug resistance.
Sawyers’ success has driven home the notion that even cells with multiple genetic flaws still rely on BCR-ABL to multiply and cause disease. If scientists can keep that enzyme in check, they can control the cancer.
Spreading success
Gleevec has helped not only CML patients, but those with other illnesses as well. The drug inhibits two tyrosine kinases in addition to BCR-ABL, and it has benefited individuals who suffer from cancers in which these other enzymes foster disease. In particular, Gleevec provides an effective therapy for patients with gastrointestinal stromal tumor (GIST) and hypereosinophilic syndrome (HES), caused by activated forms of c-Kit and PDGFR, respectively. Approximately 120,000 CML patients and 28,000 GIST patients are currently being treated with Gleevec worldwide.
Druker, Lydon, and Sawyers seized upon the known molecular defect that underlies CML, formulated the idea of tackling this root cause of the disease, crafted a specific kinase inhibitor, and designed a second-generation inhibitor when drug resistance developed. Their work has provided a model that extends well beyond CML. Now, hundreds of drugs for cancer that target specific molecules are in development and dozens have been approved. Druker, Lydon, and Sawyers have provided a stunningly successful treatment for CML and a new paradigm for cancer therapy.
by Evelyn Strauss
Key publications of Brian Druker
Oda, T., Heaney, C., Hagopian, J.R., Okuda, K., Griffin, J.D., and Druker, B.J. (1994). Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J. Biol. Chem. 269, 22925-22928.
Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., and Lydon, N.B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of BCR-ABL positive cells. Nat. Med. 2, 561-566.
Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S., and Sawyers, C.L. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344,1031- 1037.
Druker, B.J., Sawyers, C.L., Kantarjian, H., Resta, D.J., Fernandes Reese, S., Ford, J.M., Capdeville, R., and Talpaz, M. (2001). Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 344,1038-1042.
Demetri, G.D., von Mehren, M., Blanke, C.D., Van den Abbeele, A.D., Eisenberg, B., Roberts, P.J., Heinrich, M.C., Tuveson, D.A., Singer, S., Janicek, M., Fletcher, J.A., Silverman, S.G., Silberman, S.L., Capdeville, R., Kiese, B., Peng, B., Dimitrijevic, S., Druker, B.J., Corless, C., Fletcher, C.D., and Joensuu, H. (2002). Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 347, 472-480.
Druker, B.J., Guilhot, F., O’Brien, S.G., Gathmann, I., Kantarjian, H., Gattermann, N., Deininger, M.W.N., Silver, R.T., Goldman, J.M., Stone, R.M., Cervantes, F., Hochhaus, A., Powell, B.L., Gabrilove, J.L., Rousselot, P., Reiffers, J., Cornelissen, J.J., Hughes, T., Agis, H., Fischer, T., Verhoef, G., Shepherd, J., Saglio, G., Gratwohl, A., Nielsen, J.L., Radich, J.P., Simonsson, B., Taylor, K., Baccarani, M., So, C., Letvak, L., and Larson, R.A., for the IRIS Investigators. (2006). Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 355, 2408-2417.
Key publications of Nicholas Lydon
Buchdunger, E., Zimmermann, J., Mett, H., Meyer, T., Müller, M., Druker, B.J., and Lydon, N.B. (1996). Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 56,100-104.
Druker, B.J,, Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., and Lydon, N.B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of BCR-ABL positive cells. Nat. Med. 2, 561-566.
Zimmermann, J., Buchdunger, E., Mett, H., Meyer, T., and Lydon, N.B. (1997). Potent and selective inhibitors of the Abl-kinase: phenylamino-pyrimidine (PAP) derivatives. Bioog. Med. Chem. Lett. 7, 187-192.
Gambacorti-Passerini, C., le Coutre, P., Mologni, L., Fanelli, M., Bertazzoli, C., Marchesi, E., Di Nicola, M., Biondi, A., Corneo, G.M., Belotti, D., Pogliani, E., and Lydon, N.B. (1997). Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood Cells Mol. Dis. 23, 380-394.
Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S. and Sawyers, C.L. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031-1037.
Lydon, N.B. and Druker, B.J. (2004). Lessons learned from the development of imatinib. Leuk. Res. 28S1, S29-S38.
Key publications of Charles Sawyers
Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S., and Sawyers, C.L. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031-1037.
Gorre, M.E., Mohammed, M., Ellwood, K., Hsu, N., Paquette, R., Rao, P.N., and Sawyers, C.L. (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 293, 876-880.
Sawyers, C.L., Hochhaus, A., Feldman, E., Goldman, J.M., Miller, C.B., Ottmann, O.G., Schiffer, C.A., Talpaz, M., Guilhot, F., Deininger, M.W., Fischer, T., O’Brien, S.G., Stone, R.M., Gambacorti-Passerini, C.B., Russell, N.H., Reiffers, J.J., Shea, T.C., Chapuis, B., Coutre, S., Tura, S., Morra, E., Larson, R.A., Saven, A., Peschel, C., Gratwohl, A., Mandelli, F., Ben-Am, M., Gathmann, I., Capdeville, R., Paquette, R.L., and Druker, B.J. (2002). Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 99, 3530-3539.
Shah, N.P., Nicoll, J.M., Nagar, B., Gorre, M.E., Paquette, R.L., Kuriyan, J., and Sawyers, C.L. (2002). Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2, 117-125.
Shah, N.P., Tran, C., Lee, F.Y., Chen, P., Norris, D., and Sawyers, C.L. (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 305, 399-401.
Talpaz, M., Shah, N.P., Kantarjian, H., Donato, N., Nicoll, J., Paquette, R., Cortes, J., O’Brien, S., Nicaise, C., Bleickardt, E., Blackwood-Chirchir, M.A., Iyer, V., Chen, T.T., Huang, F., Decillis, A.P., and Sawyers, C.L. (2006). Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 354, 2531-2541.
Award presentation by Michael Bishop
 At the turn of the 20th century, the German medical scientist Paul Ehrlich took aim at the leading killer of his time: infections. Ehrlich was aware that some diagnostic stains for bacteria failed to react with human cells. This inspired him to imagine therapeutics that would behave in the same way — “magic bullets” as he called them, aimed exclusively at infectious agents and harmless to normal human tissues. Ehrlich’s pursuit of his vision led to the first effective treatment for syphilis, but he never found a true magic bullet. He died a disillusioned man, regarding his life in science as a failure, unconsoled by the Nobel Prize that he received in 1908. The marvel of penicillin and numerous other antibiotics that embodied Ehrlich’s vision would come later.
At the turn of the 20th century, the German medical scientist Paul Ehrlich took aim at the leading killer of his time: infections. Ehrlich was aware that some diagnostic stains for bacteria failed to react with human cells. This inspired him to imagine therapeutics that would behave in the same way — “magic bullets” as he called them, aimed exclusively at infectious agents and harmless to normal human tissues. Ehrlich’s pursuit of his vision led to the first effective treatment for syphilis, but he never found a true magic bullet. He died a disillusioned man, regarding his life in science as a failure, unconsoled by the Nobel Prize that he received in 1908. The marvel of penicillin and numerous other antibiotics that embodied Ehrlich’s vision would come later.
Although rarely mentioned, Ehrlich also imagined magic bullets for cancer. But cancer is a far more subtle adversary than infections. Cancer cells are born of normal cells, a kinship that until now has caused many of our therapies to wreak havoc with the health and welfare of cancer patients. It is fair to say that ‘chemo’ has ranked among the most dreaded of all medical procedures. And as further insult, it rarely cures. Almost a century after Paul Ehrlich, magic bullets aimed at cancer were still eluding us. The prospects improved, however, with the discovery that cancer arises from the malfunction of genes. Each malfunction sets the cancer cell apart from the normal cell; each may be a keystone for the creation of a therapeutic magic bullet. The 2009 Lasker Award for Clinical Research honors three scientists who played a central role in bringing a full-blooded magic bullet for cancer to life: Brian Druker, Nicholas Lydon, and Charles Sawyers.
Acceptance remarks

Acceptance remarks, 2009 Lasker Awards Ceremony
I would like to thank the Lasker Foundation for this prestigious honor.
I began my career in cancer research nearly 25 years ago with a dream of finding better cancer treatments. I believed the future was targeted therapies where we’d kill cancer cells without harming normal cells. To do this, I believed, we needed to figure out what was triggering the growth of cancer cells and fix it. At the time, this vision was not shared by many.
But I was fortunate; I found a collaborator, Nick Lydon, who shared my vision. The project faced many hurdles, including convincing the drug company, Novartis, to go to clinical trials. I wasn’t just a researcher, isolated in my lab, I was a doctor and I had patients who desperately needed this drug to live. In 1998, we started the clinical trials with this once-a-day pill, and within six months, every single one of our patients had their blood counts return to normal. It has been over 10 years, and today patients who once had a life expectancy of three to five years are now expected to live 30 years. With Gleevec, we’ve turned a fatal cancer into a manageable disease.
One of the best rewards of my job is every week I get to see patients who are thriving because of our work. I get to hear their stories, hear about their children and grandchildren. And I am privileged to have one of my patients here from Oregon today.
I was giving a talk recently and a woman, battling colon cancer, asked me when are we going to have a Gleevec for her cancer? I gave her a typical stilted researcher answer and told her about the 30 or more years of research that went into developing Gleevec. I went on to say that I was confident that someday we would have a Gleevec for colon cancer, but that we have to be patient. Later my wife reminded me that cancer patients don’t have the luxury of patience. As always, my wife is right. If I had been patient, I would not be standing here today. And while I — like those of you here today — want to savor the success of Gleevec, I also believe that we can’t be patient.
There is urgency to the work we do. Mary Lasker understood that urgency. She was a tireless advocate for cancer research. And I am so honored to receive this award that bears her name.
We live in a time of great promise. There are incredible opportunities in cancer research. What Gleevec tells us is that by understanding cancer we can develop effective treatments. Gleevec tells us we are on the right track, but we can’t be complacent. We can’t be patient. We must seize this momentum to reach the finish line of curing cancer. Thank you for this great honor.

Acceptance remarks, 2009 Lasker Awards Ceremony
It is an enormous honor and pleasure to be here with my co-recipients to receive the Lasker~DeBakey Award.
Like many pharmaceutical discoveries, imatinib was a team effort that was built on a foundation of basic science discoveries, some of which have previously been recognized by the Lasker Foundation. I am honored to accept this award on behalf of myself and my collaborators at Ciba-Geigy, who contributed to the development of imatinib.
My interest in kinases started during my postgraduate days at the University of Dundee, where I was profoundly influenced by the work of Philip Cohen’s lab on the regulation of glycogen metabolism by reversible protein phosphorylation. Armed with a growing interest in protein kinases, I was fortunate to move into applied research at a time when pioneering studies in the oncogene field had come together to clearly implicate the Bcr-Abl kinases in the pathogenesis of CML. In retrospect, being new to the pharmaceutical industry was a big advantage, as I was naive to the complex path a drug discovery idea must take from its inception in the lab through to the clinic. Luckily, I was not alone in this journey and had the fortune of working with outstanding colleagues and mentors at Ciba-Geigy and collaborating with Brian Druker in the translation of imatinib into the clinic.
I consider myself extremely blessed in having been able to contribute to a project that has made a difference to cancer patients. I thank you for this great honor and hope that it will motivate young scientists to work on translating advances that have been made in basic research into treatments that make an impact on patients’ lives.
Acceptance remarks, 2009 Lasker Awards Ceremony
It is a humbling and unexpected honor to be here accepting the Lasker~DeBakey Clinical Research Award. Brian Druker and I met nearly 20 years ago when we were both postdoctoral fellows studying the signaling properties of the BCR-ABL enzyme. Brian was in Boston, I was in Los Angeles. I met Nick Lydon a few years later in Basel, Switzerland, when he and Brian invited me to join the effort to move the compound that would become Gleevec into the clinic. Successful partnerships are born in unpredictable ways. Who knew what would follow?
Among our trio, I am known primarily as the resistance guy. In 2001 my group discovered that patients who relapse while taking Gleevec have mutations in the BCR-ABL enzyme that prevent the drug from binding its target. This was a big surprise because all bets were on another explanation — that tumors would escape their need for BCR-ABL because they had so many other genetic mutations that could presumably drive the growth of the tumor. Instead, the answer was elegantly simple. The tumor does everything in its power to maintain BCR-ABL activity. In hindsight, this was clear evidence of a general principle we now call oncogene addiction that guides much of cancer drug development today.
Many people played a role in the remarkable convergence of events that allowed us to move so quickly from discovering the cause of resistance in 2001 to a solution to overcome it just a few years later. Pasteur said that “chance favors the prepared mind.” I credit Owen Witte, my postdoctoral mentor, for ‘preparing’ my mind to connect the various pieces of the resistance puzzle that we faced first in the clinic, then in the laboratory. Mercedes Gorre and Neil Shah, who are both here today, conducted the experiments in my lab that led to the discovery of the resistance mutations and courageously persevered when their initial results were challenged. John Kuriyan solved the structure of Gleevec bound to ABL, then partnered with us to understand how the many different mutations we were finding in patients confer resistance. Only through that collaboration did we come to the hypothesis that an inhibitor that binds BCR-ABL differently might offer a common solution against the many different Gleevec-resistant mutations. In an amazing coincidence, Frank Lee and Rob Kramer at Bristol Myers Squibb had a compound with precisely the properties we were imagining but that they had originally discovered for a different purpose. Four years later that compound, dasatinib or Sprycel, was approved by the FDA for Gleevec-resistant chronic myeloid leukemia.
In closing, I thank the patients who participated in these clinical trials, many of whom have sent me messages of joy ever since the Lasker announcement. I also thank my parents, my brother and sister, and my wife and children, all here today, for supporting my dedication to this work.
Interview with Brian J. Druker, Nicholas B. Lydon, and Charles L. Sawyers
Video Credit: Susan Hadary
