
Ralf F.W. Bartenschlager
Heidelberg University

Charles M. Rice
Rockefeller University

Michael J. Sofia
Arbutus Biopharma
The 2016 Lasker~DeBakey Clinical Medical Research Award honors three scientists who developed a system to study the replication of the virus that causes hepatitis C and used this system to revolutionize the treatment of this chronic, often lethal disease. For more than a decade, Ralf F. W. Bartenschlager (University of Heidelberg) and Charles M. Rice (Rockefeller University) attempted to coax the hepatitis C virus (HCV) to multiply inside lab-grown host cells. They conquered one challenge after another and eventually triumphed. With similar perseverance and imagination, Michael J. Sofia (formerly at Pharmasset; now at Arbutus Biopharma) then exploited this system to test and invent candidate drugs, whose novel design allowed targeting of the liver, where HCV resides, and creation of a medicine with unprecedented potency and safety.
Reaching consensus
To study a virus and develop drugs against it, scientists need a way to grow it in the lab. With the isolation of HCV in 1989, Michael Houghton (Albert Lasker Clinical Medical Research Award, 2000) opened a new avenue toward that end. Investigators could use standard recombinant DNA techniques to produce the virus’s RNA. After putting it into cells, they expected that the host machinery would use this genetic code to construct infectious HCV.
The approach flopped.
As researchers tried to prod HCV to replicate in cells, they began wondering whether they had captured its entire sequence. Knowing that viral-preparation methods could introduce errors, Rice (then at Washington University) and postdoctoral fellow Alexander Kolykhalov deployed a method for defining the complete HCV RNA that would avoid these technical pitfalls.
In 1996, their strategy revealed an unanticipated structure at the end of the virus. This finding, also reported by Kunitada Shimotohno (then at the National Cancer Center Research Institute, Tokyo), implied that all of the constructed genomes had failed to replicate in the lab because they lacked this crucial feature.
With high hopes, Rice made a series of HCV sequences that included the bona fide end and injected them into chimpanzees. Unfortunately, these RNAs produced no hepatitis or other signs of infection.
Undaunted, he performed further analysis and found that the RNAs had acquired sequence changes—either while being amplified in the lab or before that, when the virus was replicating inside an infected human. These genetic errors, he reasoned, weakened HCV’s ability to propagate. He created a “consensus” genome—one that, at every position, held the most common RNA letter rather than a deleterious one.
The resulting HCV RNA infected chimps and gave them hepatitis, Rice and Jens Bukh (National Institutes of Health) independently reported in 1997. Researchers had designed and produced a functional virus in the lab. Surely the consensus RNAs would generate HCV in lab-grown cells and thus allow investigators to dissect its detailed biology.
Replicating success
In the meantime, Bartenschlager (then at the University of Mainz) had made a different consensus HCV sequence. He introduced it into various host liver cells, but never detected replication. Rice’s consensus RNA also flunked this test.
Bartenschlager had an idea. He knew that relatives of HCV could lose a chunk of their genomes—the region that encodes viral-packaging proteins—yet still multiply inside host cells. He wondered whether he could replace these dispensable sequences with a genetic marker that would expose cells that contain replicating HCV.
In the late 1990’s, he and postdoctoral fellow Volker Lohmann gutted their consensus RNA and inserted a gene whose product confers resistance to a lethal drug. If the HCV RNA-copying machinery did its job, it would amplify not only viral RNA, but also that of the drug-resistance gene. Consequently, host cells that carried this “replicon” would withstand the otherwise deadly toxin.
The scheme worked. The ability of the replicons to bestow a survival advantage upon host liver cells had thus allowed researchers to detect HCV replication in the lab. Still, as they clinked in celebration, their brows wrinkled. Surviving cells contained thousands of RNAs, but only about one in a million cells that received the input RNA survived.
The replicons that flourished, it turned out, had picked up adaptive sequence changes inside cells, Rice and Bartenschlager independently showed. After the investigators engineered these replication-enhancing mutations back into the starting RNA, production of infected cells jumped 500- to 10,000-fold.
For the first time, researchers had generated efficient HCV replication in the lab. Because the replicons did not produce infectious virus particles, the system could be used safely without high-level precautions. Furthermore, the mini-genomes encoded proteins critical for HCV multiplication, important drug targets.
Unmasking a champion
The pharmaceutical industry now had a manageable way to test whether candidate agents thwart HCV inside living liver cells. New medicines were sorely needed. Standard regimens required weekly injections of interferon, which delivers severe side effects. Furthermore, treatment took 24-72 weeks and frequently failed.
In their quest to improve therapy, scientists at Pharmasset, a small biotech company founded by Raymond Schinazi, focused on HCV’s RNA-copying enzyme. Its active site was similar among disparate types of HCV and the enzyme has no human counterpart, so perhaps the team could develop an inhibitor that would combat multiple HCV genotypes without disrupting host physiology.
The investigators set their sights on chemicals that resemble normal RNA building blocks, but differ in a crucial way. These so-called nucleoside analogs attach to a growing RNA chain, but when the copying enzyme tries to add the next subunit, it can’t. The analogs’ extra chemical groups interfere, and RNA elongation stops.
In 2005, Jeremy L. Clark, a member of the Pharmasset team, identified a nucleoside analog that blocks HCV replication in the replicon system. In people, its safety profile looked promising, but most of it broke down into an inactive form.
By modifying its chemical properties, the Pharmasset scientists, now under the direction of Michael Sofia, solved these problems. In 2010, they reported that their improved nucleoside analog slashes viral load when combined with a different class of drug that inhibits a different HCV enzyme, but large and frequent doses of the analog were required.
Aiming to boost its potency, Sofia hit on an unconventional idea. The team’s studies had revealed that the original analog might not be a dead-end after all. Through a series of reactions, enzymes in the body convert a small proportion of it into a different compound that foils HCV replication and persists intact in liver cells.
To harness the potential of this powerful and stable inhibitor, Sofia had to overcome a substantial challenge. He needed to supply not the original analog, which is transformed inefficiently into the desired chemical, but a specific molecular relative. This relative carries a phosphate chemical group, whose negative charge renders it unable to traverse oily cell membranes.
Sofia and his colleagues aimed to mask the phosphate so the compound would slip into cells. In his dream scenario, liver enzymes would then disrobe the agent and, with its charged portion revealed, it would be stuck. Conversion enzymes would set upon it and turn it into the active drug. His vision also promised to minimize harmful effects that might result from delivery to other parts of the body. Only liver cells possess the natural metabolic capabilities that trap the compound inside.
With intense tweaking and evaluation, Sofia designed a chemical candidate that performed well in the replicon system and passed other tests. Early clinical trials showed extremely promising results, and Gilead Sciences acquired Pharmasset in early 2012. In January 2013, the Pharmasset group, Gilead, and clinical collaborators from New Zealand published the dramatic findings. In combination with ribavirin, a toxic but non-interferon-based antiviral agent, the new drug, sofosbuvir, eradicated HCV long term. In people infected with some viral genotypes, no virus could be detected 24 weeks after a 12-week treatment period ended. The absence of interferon in this regimen launched a new era in curative HCV therapy.
On December 6, 2013, a sofosbuvir (Sovaldi®) regimen was approved by the US Federal Drug Administration (FDA) for some HCV genotypes. Finally, people with chronic HCV infections had an interferon-free therapy. Subsequently, sofosbuvir proved effective in diverse patient populations and across viral genotypes.
In the meantime, the replicon assay was fueling other scientists’ discovery of new drugs. A Bristol-Myers Squibb group led by Min Gao identified a compound that targets an HCV protein of unknown function. This achievement showcased another strength of the replicon system—its ability to let the cell reveal what is important for replication, regardless whether scientists understand the underpinnings.
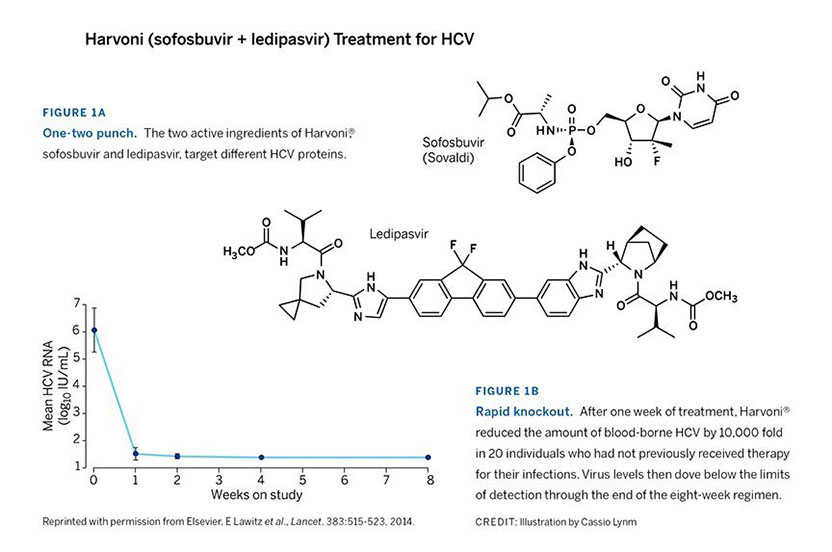
Gilead rapidly developed a derivative of the compound Gao and colleagues had pioneered. The combination of Gilead’s new agent, ledipasvir, plus sofosbuvir (Figure 1A) rapidly quashes the virus. This regimen boasts cure rates of 94-99% in only 8-12 weeks of therapy (Figure 1B), even among difficult-to-treat patients. The ledipasvir/sofosbuvir combination, Harvoni®, is now FDA-approved for numerous types of HCV infection, including the most common form in the US and Europe. It is the first HCV treatment that avoids both interferon and ribavirin. Four other such medications have since been approved. Sofosbuvir serves as the backbone of two, and the other two are based on different compounds.
Bartenschlager, Rice, and Sofia surmounted numerous hurdles as they devised innovative solutions to the biological and chemical obstacles that confronted them. Their victories culminated in a safe, effective, oral therapy for HCV that set a new standard and transformed the treatment of a devastating illness.
by Evelyn Strauss
Key Publications of Ralf F.W. Bartenschlager
Lohmann, V., Körner, F., Koch, J.O., Herian, U., Theilmann, L., and Bartenschlager, R. (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 285, 110-113.
Bartenschlager, R. and Lohmann, V. (2000). Replication of hepatitis C virus. J. Gen. Virol. 81, 1631-1648.
Krieger, N., Lohmann, V., and Bartenschlager, R. (2001). Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75, 4614-4624.
Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z., Murthy, K., Habermann, A., Kräusslich, H.-G., Mizokami, M., Bartenschlager, R., and Liang, T.J. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791-796.
Pietschmann, T., Kaul, A., Koutsoudakis, G., Shavinskaya, A., Kallis, S., Steinmann, E., Abid, K., Negro, F., Dreux, M., Cosset, F.L., and Bartenschlager, R. (2006). Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA. 103, 7408-7413.
Key Publications of Charles M. Rice
Grakoui, A., Lin, C., Wychowski, C., Feinstone, S., and Rice, C.M. (1993). Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67, 1385-1395.
Kolykhalov, A.A., Feinstone, S.M., and Rice, C.M. (1996). Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 70, 3363-3371.
Kolykhalov, A.A., Agapov, E.V., Blight, K.J., Mihalik, K., Feinstone, S.M., and Rice, C.M. (1997). Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 277, 570-574.
Blight, K.J., Kolykhalov, A.A., and Rice, C.M. (2000). Efficient initiation of HCV RNA replication in cell culture. Science. 290, 1972-1974.
Lindenbach, B.D., Evans, M.J., Syder, A.J., Wölk, B., Tellinghuisen, T.L., Liu, C.C., Maruyama, T., Hynes, R.O., Burton, D.R., McKeating, J.A., and Rice, C.M. (2005). Complete replication of hepatitis C virus in cell culture. Science. 309, 623-626.
Key Publications of Michael J. Sofia
Sofia, M.J., Bao, D., Chang, W., Du, J., Nagarathnam, D., Rachakonda, S., Reddy, P.G., Ross, B.S., Wang, P., Zhang, H.R., Bansal, S., Espiritu, C., Keilman, M., Lam, A.M., Steuer, H.M., Niu, C., Otto, M.J., and Furman, P.A. (2010). Discovery of a β-ᴅ-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53, 7202-7218.
Sofia, M.J., Furman, P.A., and Symonds, W.T. (2010). 2′-F-2′-C-methyl nucleosides and nucleotides for the treatment of hepatitis C virus: from discovery to the clinic. In: Accounts in Drug Discovery: Case Studies in Medicinal Chemistry. Chapter 11. Royal Society of Chemistry, London, pp. 238-266.
Sofia, M.J. (2013). Nucleotide prodrugs for the treatment of HCV infection. Adv. Pharmacol. 67, 39-73.
Sofia, M.J. (2014). Beyond sofosbuvir: what opportunity exists for a better nucleoside/nucleotide to treat hepatitis C? Antiviral Res. 107, 119-124.
Sofia, M.J. (2015). Sofosbuvir: a breakthrough curative therapy for the treatment of HCV infection. In: Medicinal Chemistry Reviews. Vol. 50. Med. Chem. Div., Am. Chem. Soc., Foster City, CA. pp. 397-416.
Editorial Comments on Therapy for Hepatitis C
Hoofnagle, J.H. and Sherker, A.H. (2014). Therapy for Hepatitis C – the costs of success. New Engl. J. Med. 370, 1552-1553.
Liang, T.J. and Ghany, M.G. (2014). Therapy of Hepatitis C – back to the future. New Engl. J. Med. 370, 2043-2047.
Award presentation by Harold Varmus
Most of us don’t spend much time thinking about our livers. Unlike our pumping hearts and our reasoning brains, the liver is normally a silent, though essential, partner. Now it is quietly working on our lunch, keeping us in metabolic balance. But when damaged by alcohol, infiltrated by metastatic cancer, or felled by a viral infection, it can no longer be ignored.
Hepatitis C virus (or HCV) is one of the things that can bring the liver to our attention. The virus is growing persistently in the livers of an estimated 170 million people worldwide and over 3 million in the US. With time, HCV can destroy the liver’s essential functions, since an infection is often a prelude to cirrhosis or liver cancer.
Acceptance remarks
Acceptance remarks, 2016 Lasker Awards Ceremony
Passion, most often requiring an adequate trigger, supporting people, and luck are the most important ingredients for a scientific career. I had the fortune to have all of them.
When I grew up with my grandparents in an industrial suburb of Mannheim, my main interest was playing soccer and I had no clue what my future profession would be. This changed in high school where I met an excellent teacher who raised my interest in biology. Recognizing my new inspiration, my grandparents bought me a small microscope as a Christmas present that I used in my spare time to examine all sorts of biological samples, although often with disappointing results because of its poor resolution.
During those days I decided to study biology even though people were telling me at that time, the late 1970s, that the demand for biologists in Germany is very low; one biologist per year, so why should I be this person? Nevertheless, my passion for this topic was stronger than any concerns about my professional future, so after a 4-year interim time during which I was trained and practiced as a police officer, I began to study biology at Heidelberg University.
During my time as student, I developed a growing interest in molecular biology, which in Germany was at its infancy. Fortunately, there were several professors in Heidelberg, who did postdocs in the US and after their return to Germany started to build up labs that used most modern molecular biology methodologies. One of these labs was headed by Heinz Schaller. Heinz worked on hepatitis B virus, a topic that I found fascinating, but getting access to his lab was not an easy task. resistance. Fortunately, he accepted me as lab practical student to establish a method for cloning duck hepatitis B virus mutants. I assume I did my job right, because Heinz took me under his wing and really taught me the secrets of scientific working. I left his lab in 1991, and in 2002 I got the chance to return back to Heidelberg to set up a new research department made possible by a foundation set up by Heinz and his wife Chica.
Success in science also requires luck. My biggest luck was the privilege to host numerous incredibly talented students and postdocs in my lab. While all have contributed tremendously, there was one person who influenced my career most: Volker Lohmann, my very first PhD student. Recruiting Volker was one of the biggest strokes of luck in my scientific life as he was not only the key person to set up the first HCV cell culture system, but also an exceptional mind creating true team spirit and synergy within the research team.
Being a scientist is not just a job, it is a vocation—the pursuit of knowledge and the elation of discovery are the twin triggers of passion in science. But this vocation also brings long working hours, hectic days, and frequent travel. Without the continuous encouragement of my wife Judith, and without the enrichment that my children Marie, Nora, and Lorenz have provided to my life, I would not be here today. Thank you.
Passion, most often requiring an adequate trigger, supporting people, and luck are the most important ingredients for a scientific career. I had the fortune to have all of them.
When I grew up with my grandparents in an industrial suburb of Mannheim, my main interest was playing soccer and I had no clue what my future profession would be. This changed in high school where I met an excellent teacher who raised my interest in biology. Recognizing my new inspiration, my grandparents bought me a small microscope as a Christmas present that I used in my spare time to examine all sorts of biological samples, although often with disappointing results because of its poor resolution.
During those days I decided to study biology even though people were telling me at that time, the late 1970s, that the demand for biologists in Germany is very low; one biologist per year, so why should I be this person? Nevertheless, my passion for this topic was stronger than any concerns about my professional future, so after a 4-year interim time during which I was trained and practiced as a police officer, I began to study biology at Heidelberg University.
During my time as student, I developed a growing interest in molecular biology, which in Germany was at its infancy. Fortunately, there were several professors in Heidelberg, who did postdocs in the US and after their return to Germany started to build up labs that used most modern molecular biology methodologies. One of these labs was headed by Heinz Schaller. Heinz worked on hepatitis B virus, a topic that I found fascinating, but getting access to his lab was not an easy task. resistance. Fortunately, he accepted me as lab practical student to establish a method for cloning duck hepatitis B virus mutants. I assume I did my job right, because Heinz took me under his wing and really taught me the secrets of scientific working. I left his lab in 1991, and in 2002 I got the chance to return back to Heidelberg to set up a new research department made possible by a foundation set up by Heinz and his wife Chica.
Success in science also requires luck. My biggest luck was the privilege to host numerous incredibly talented students and postdocs in my lab. While all have contributed tremendously, there was one person who influenced my career most: Volker Lohmann, my very first PhD student. Recruiting Volker was one of the biggest strokes of luck in my scientific life as he was not only the key person to set up the first HCV cell culture system, but also an exceptional mind creating true team spirit and synergy within the research team.
Being a scientist is not just a job, it is a vocation—the pursuit of knowledge and the elation of discovery are the twin triggers of passion in science. But this vocation also brings long working hours, hectic days, and frequent travel. Without the continuous encouragement of my wife Judith, and without the enrichment that my children Marie, Nora, and Lorenz have provided to my life, I would not be here today. Thank you.
Acceptance remarks, 2016 Lasker Awards Ceremony
As I look back over nearly 40 years of studying viruses, I never imagined that I would be standing here today. Viruses are great teachers, and unraveling their secrets continues to reveal new wonders about biology and evolution.
My interest in flaviviruses was kindled as a postdoc and continued when I began my laboratory at Washington University. We studied the prototype family member, yellow fever virus, a nasty mosquito-borne virus made famous by Walter Reed. It became the poster child for human vaccination based on a live-attenuated variant derived in 1937 by Nobel Prize recipient Max Theiler.
In the midst of exploring the details of yellow fever replication, hepatitis C appeared on the scene in 1989 as the major cause of non-A, non-B hepatitis. Given its similarity to yellow fever and other flaviviruses, we began a small effort on this new human virus associated with progressive liver disease and cancer. We weren’t specifically searching for a cure. We were just trying to better understand the virus. Together with Ralf and others in the field, we made progress but struggled to get the virus to grow in the lab. With each apparent breakthrough, we were met with new roadblocks to overcome. In both patients and in the laboratory hepatitis C was a persistent troublemaker.
I must admit that most of us in the field became increasingly frustrated by the difficulty in working with this virus and our slow progress at a time when new therapies were sorely needed by patients. But when the breakthroughs finally came, the tools were in hand to validate potential antiviral targets and screen for inhibitory compounds. A flood of new drugs rapidly ensued. Regimens with amazing efficacy and very few side effects are now approved and in the clinic, although we still face many public health challenges for global implementation.
It is wonderful to have played a small role in this biomedical success story. About 20 years ago, I was contacted by a family whose little girl had hepatitis C. For years, they agonized over whether to treat or not. They decided to wait for the new drugs. Last week, this message arrived: “Hi Charlie. I just read that you will be receiving a Lasker Award for your work in helping cure Hep C. Congratulations!!! We are so excited for you and remain grateful that our daughter is cured and leading a happy normal life. In fact, happily married with an eighteen month old, our daughter is now expecting her second child.” For a basic scientist, no reward is more unexpected or better than this.
In closing, I remember my mother asking “why do you work on yellow fever—we have a vaccine for that, don’t we?” She was right of course, but we were curious about that virus and forged ahead anyway. This experience with yellow fever paved the way for our work on hepatitis C and to me reinforces the importance of keeping a diverse research portfolio—you never know what’s coming next! It also underscores the importance of supporting and promoting fundamental curiosity-driven research as the driver of great medical advances.
Acceptance remarks, 2016 Lasker Awards Ceremony
As I look back over nearly 40 years of studying viruses, I never imagined that I would be standing here today. Viruses are great teachers, and unraveling their secrets continues to reveal new wonders about biology and evolution.
My interest in flaviviruses was kindled as a postdoc and continued when I began my laboratory at Washington University. We studied the prototype family member, yellow fever virus, a nasty mosquito-borne virus made famous by Walter Reed. It became the poster child for human vaccination based on a live-attenuated variant derived in 1937 by Nobel Prize recipient Max Theiler.
In the midst of exploring the details of yellow fever replication, hepatitis C appeared on the scene in 1989 as the major cause of non-A, non-B hepatitis. Given its similarity to yellow fever and other flaviviruses, we began a small effort on this new human virus associated with progressive liver disease and cancer. We weren’t specifically searching for a cure. We were just trying to better understand the virus. Together with Ralf and others in the field, we made progress but struggled to get the virus to grow in the lab. With each apparent breakthrough, we were met with new roadblocks to overcome. In both patients and in the laboratory hepatitis C was a persistent troublemaker.
I must admit that most of us in the field became increasingly frustrated by the difficulty in working with this virus and our slow progress at a time when new therapies were sorely needed by patients. But when the breakthroughs finally came, the tools were in hand to validate potential antiviral targets and screen for inhibitory compounds. A flood of new drugs rapidly ensued. Regimens with amazing efficacy and very few side effects are now approved and in the clinic, although we still face many public health challenges for global implementation.
It is wonderful to have played a small role in this biomedical success story. About 20 years ago, I was contacted by a family whose little girl had hepatitis C. For years, they agonized over whether to treat or not. They decided to wait for the new drugs. Last week, this message arrived: “Hi Charlie. I just read that you will be receiving a Lasker Award for your work in helping cure Hep C. Congratulations!!! We are so excited for you and remain grateful that our daughter is cured and leading a happy normal life. In fact, happily married with an eighteen month old, our daughter is now expecting her second child.” For a basic scientist, no reward is more unexpected or better than this.
In closing, I remember my mother asking “why do you work on yellow fever—we have a vaccine for that, don’t we?” She was right of course, but we were curious about that virus and forged ahead anyway. This experience with yellow fever paved the way for our work on hepatitis C and to me reinforces the importance of keeping a diverse research portfolio—you never know what’s coming next! It also underscores the importance of supporting and promoting fundamental curiosity-driven research as the driver of great medical advances.
The 2016 Clinical Award video
Video Credit: Flora Lichtman
